Resources

From Data to Insight – A Practical Introduction to Interpreting Malaria Genetics for Surveillance
We are excited to announce a free, self-paced online course offered by PlasmoGenEpi. The course is intended for researchers, students and public health officials who are new to the field of malaria genetic epidemiology. It covers foundational concepts through video lectures, interactive case studies, quizzes, job-aids, and moderated discussion forums. Learn more about the course and enroll here https://www.plasmogenepi.org/OnlineCourse

An Illumina-compatible, modular, highly multiplexed amplicon sequencing panel P falciparum with a broad range of targets and the ability to genotype low density, polyclonal field samples. Two modules:
1) a diversity panel with 165 highly diverse microhaplotypes
2) 118 targets that cover drug and diagnostic resistance genes as well as non-falciparum species identification and vaccine candidates.
dcifer- Distance for Complex Infections: Fast Estimation of Relatedness
An IBD-based method to calculate genetic distance between polyclonal infections by estimating relatedness from biallelic and multiallelic data. In addition to estimates, the package provides a likelihood function and statistical inference. Functions for reading and reformatting data, performing preparatory steps, and visualizing the results are also included.

moire- Multiplicity Of Infection and allele frequency REcovery from noisy polyallelic data
A package implementing an MCMC based approach to estimating complexity of infection and population allele frequencies from polyallelic genomics data.
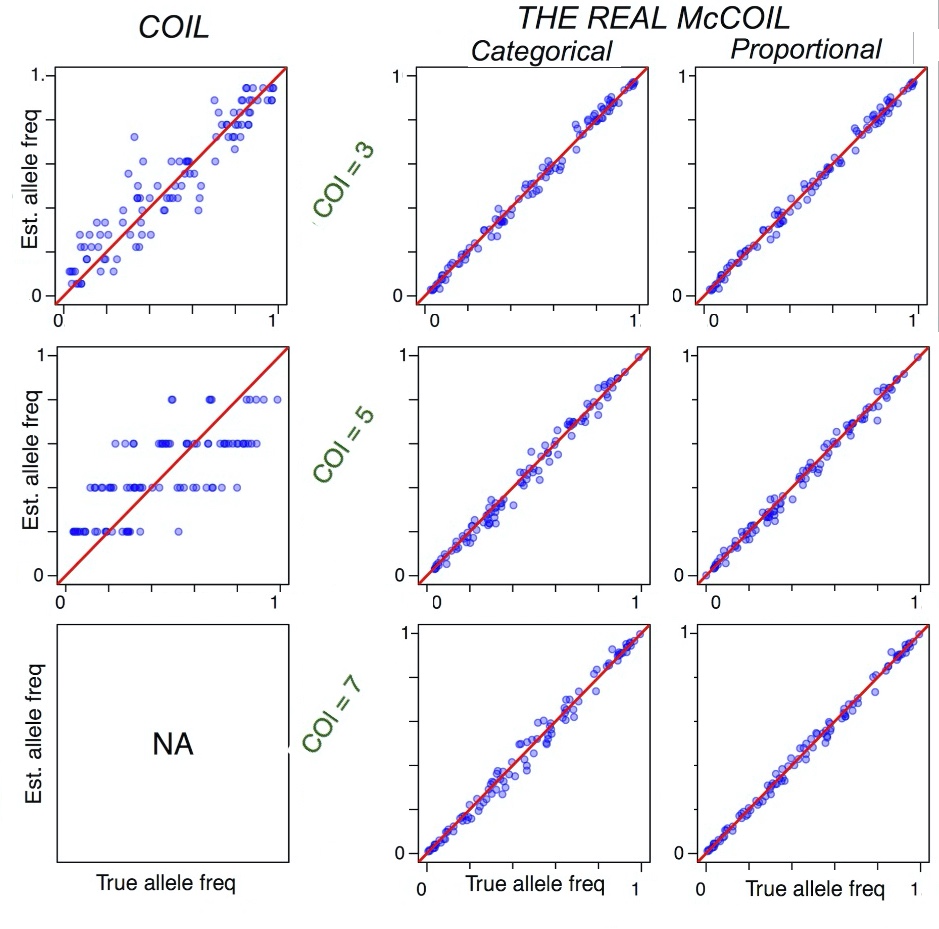
A Markov chain Monte Carlo method that estimates complexity of infection and population allele frequencies using SNP data obtained from Sequenom or similar types of SNP assays. We used the data in two ways:
categorical- where we consider SNP to be heterozygous or homozygous
proportional- where relative signal intensity for each allele is used
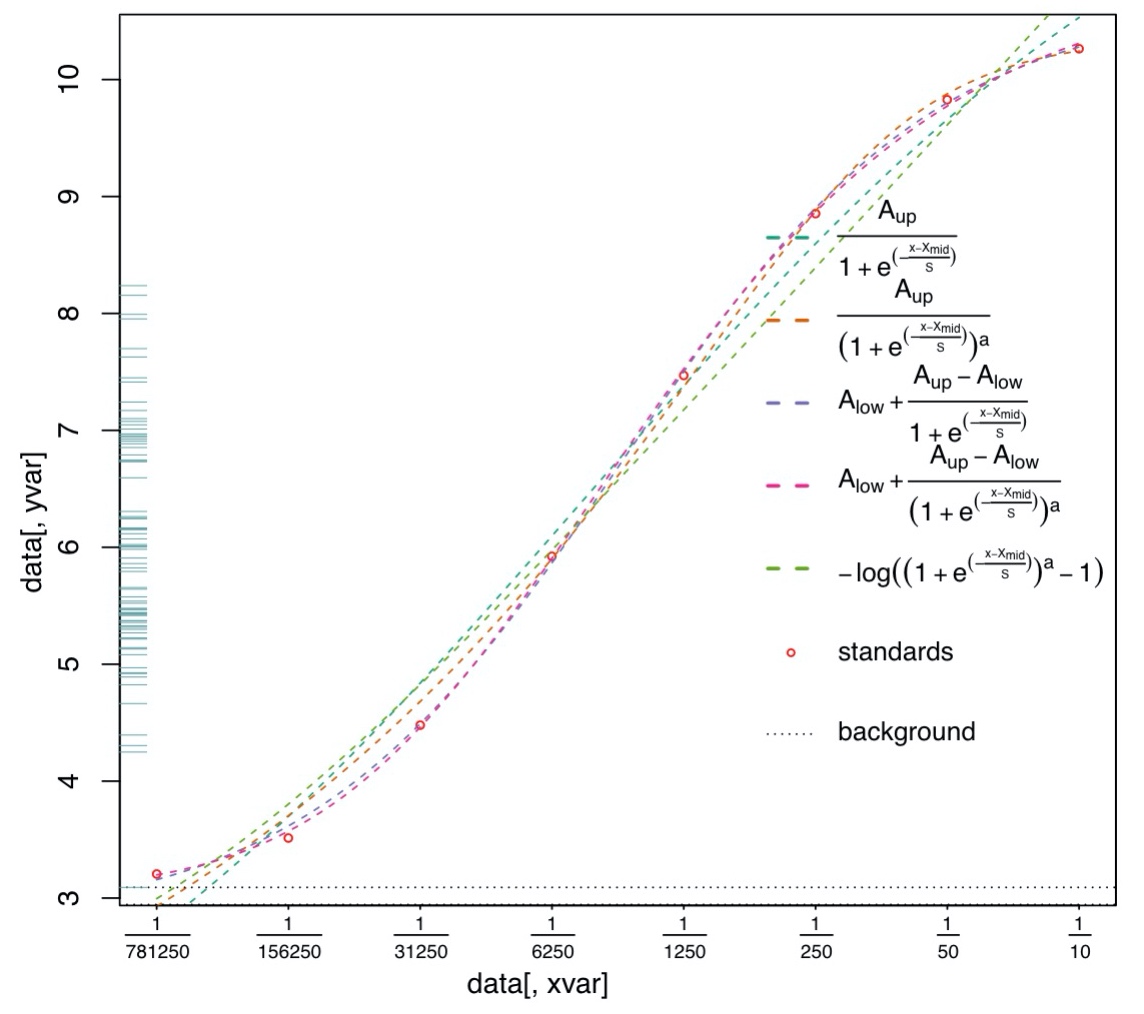
flexfit- Flexible format standard curve fitting & data processing
Flexible format standard curve fitting and data processing (R package)
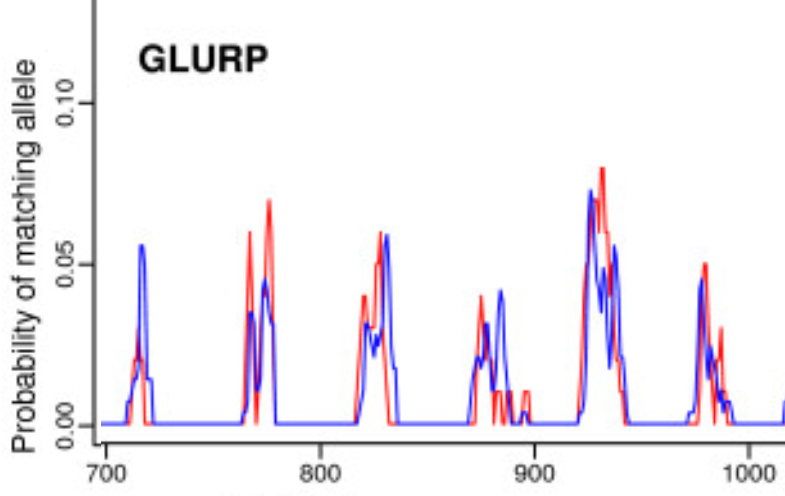
MicroSPAT- MicroSatellite Parameterized Analysis Tools
A collection of tools for semi-automated analysis of raw capillary electrophoresis (CE) data output by the ABI 3730, and has been designed specifically for working with multi-allelic samples. Developed to improve reproducibility and consistency of microsatellite analysis between analysts.

Navigating a fast paced lab is challenging. This is how we do it.
Data repositories:

ClinEpiDB- Clinical Epidemiology Database -PRISM Data
Data from our longitudinal cohorts of P. falciuparum infection in Uganda (PRISM 1 and PRISM 2) is available on this open-access data analysis platform.